
Hypertrophic Cardiomyopathy (HCM) Mouse Model: Mybpc3 KO
Hypertrophic Cardiomyopathy Mouse Model: C57BL/6JGpt-Mybpc3em19Cd10922/Gpt
MYBPC3 encodes cardiac myosin-binding protein C (cMyBP-C), a thick filament-associated protein critical for regulating myocardial contraction and relaxation by modulating actin-myosin cross-bridge interactions. It acts as a "brake" to limit excessive cross-bridge cycling, maintaining sarcomere structural integrity and calcium sensitivity. MYBPC3 knock-out (KO) mice exhibit eccentric hypertrophic cardiomyopathy characterized by left ventricular dilation, reduced systolic function, and electrocardiographic abnormalities (e.g., ST-segment elevation). These mice also demonstrate accelerated myosin cycling kinetics and increased arrhythmia susceptibility, mirroring human MYBPC3-related cardiomyopathy pathology.
Strategy
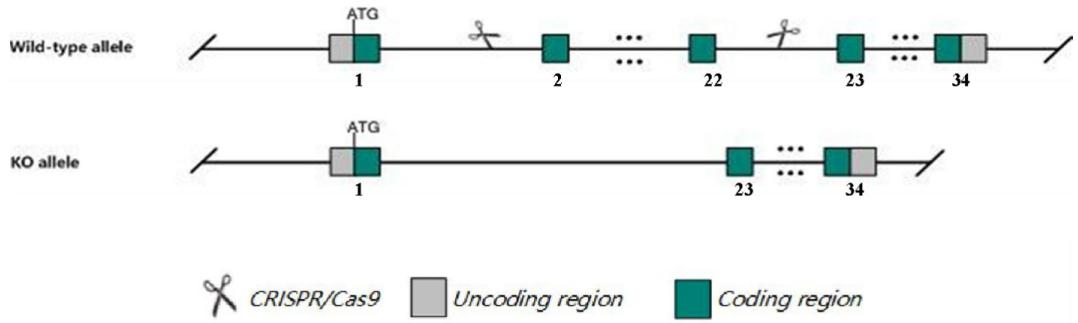
Fig.1 Schematic diagram of C57BL/6JGpt-Mybpc3em19Cd10922/Gpt model strategy.
The ablation of MYBPC3 in C57BL/6JGpt-Mybpc3em19Cd10922/Gpt mice.
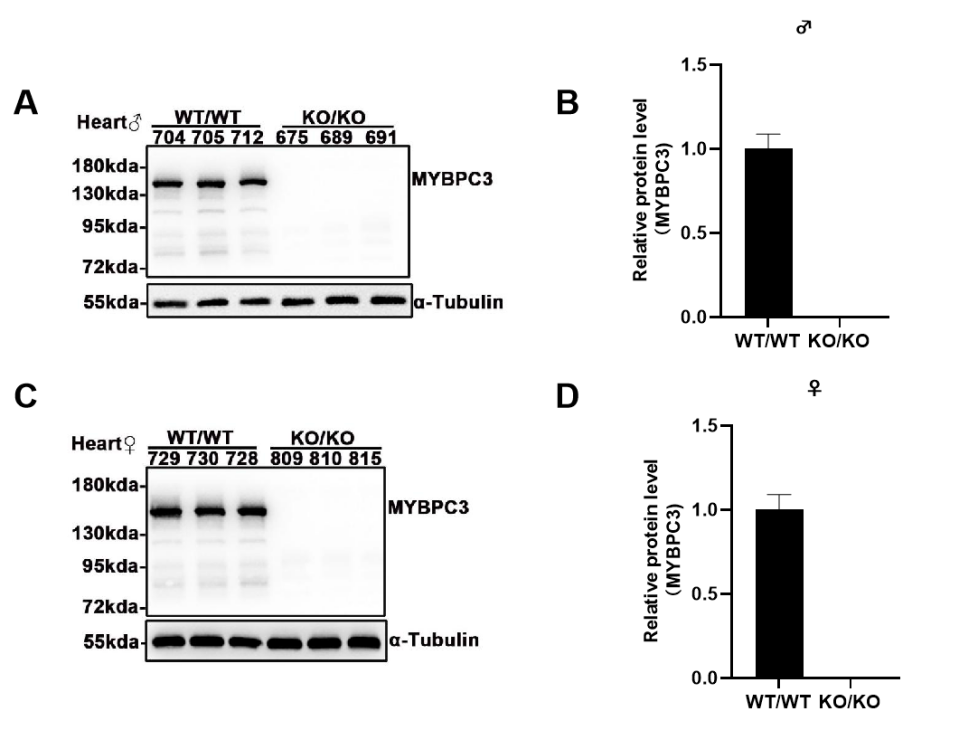
Fig 2. MYBPC3 protein is not expressed in C57BL/6JGpt-Mybpc3em19Cd10922/Gpt mice.
Western blot analysis using a monoclonal antibody specific to murine MYBPC3 confirmed the genotype-dependent expression profile in cardiac tissues. Cardiac lysates from WT controls exhibited robust MYBPC3 expression (n=6, 3 males and 3 females), whereas the C57BL/6JGpt-Mybpc3em19Cd10922/Gpt cohort demonstrated complete loss of MYBPC3 protein (n=6, 3 males and 3 females). No sex-specific differences in expression levels were observed within groups. Antibody specificity was validated by the absence of nonspecific bands in myocardial lysates from knockout animals, corroborating the genetic ablation of Mybpc3 in this model.
C57BL/6JGpt-Mybpc3em19Cd10922/Gpt mice display eccentric hypertrophic cardiomyopathy at 14 days of age.
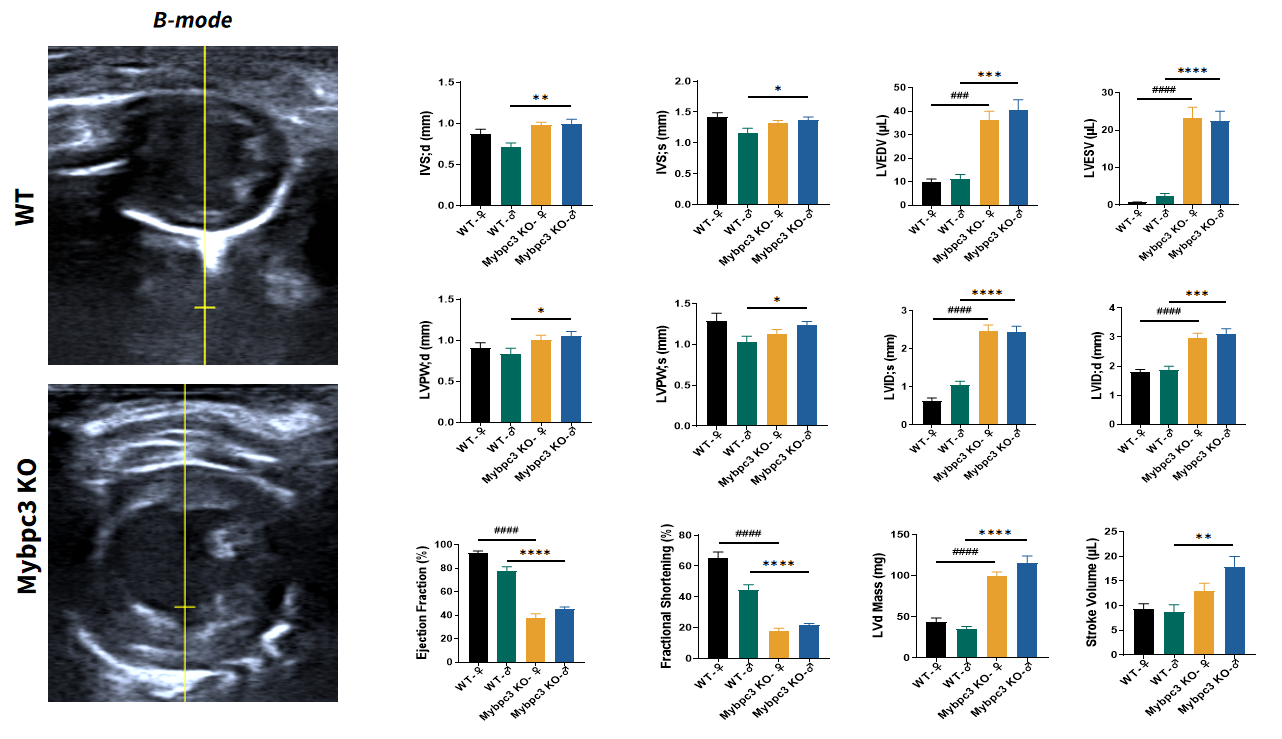
Fig 3. Echocardiography of C57BL/6JGpt-Mybpc3em19Cd10922/Gpt mice at P14.
Echocardiographic analysis revealed significant cardiac enlargement in C57BL/6JGpt-Mybpc3em19Cd10922/Gpt mice compared to wild-type (WT) controls. The mutant hearts demonstrated significant increases in both interventricular septal thickness at end-diastole (IVSd) and left ventricular posterior wall thickness at end-diastole (LVPWd), consistent with myocardial hypertrophy. Concurrently, pathological elevations in left ventricular end-diastolic volume (LVEDV) and end-systolic volume (LVESV), coupled with reduced ejection fraction (EF) and fractional shortening (FS), collectively indicated ventricular dilation and systolic impairment. These structural and functional aberrations establish that Mybpc3 deficiency is causally linked to the development of eccentric hypertrophic cardiomyopathy during the adolescent stage, recapitulating features of human MYBPC3-associated cardiomyopathy. n=10 per group, all data represented as MEAN ± SEM.****P<0.0001,by t-test. n=10 per group, all data represented as MEAN ± SEM.**P<0.01, ***P<0.001, ****P<0.0001,by t-test.
Cardiac remolding in C57BL/6JGpt-Mybpc3em19Cd10922/Gpt mice.
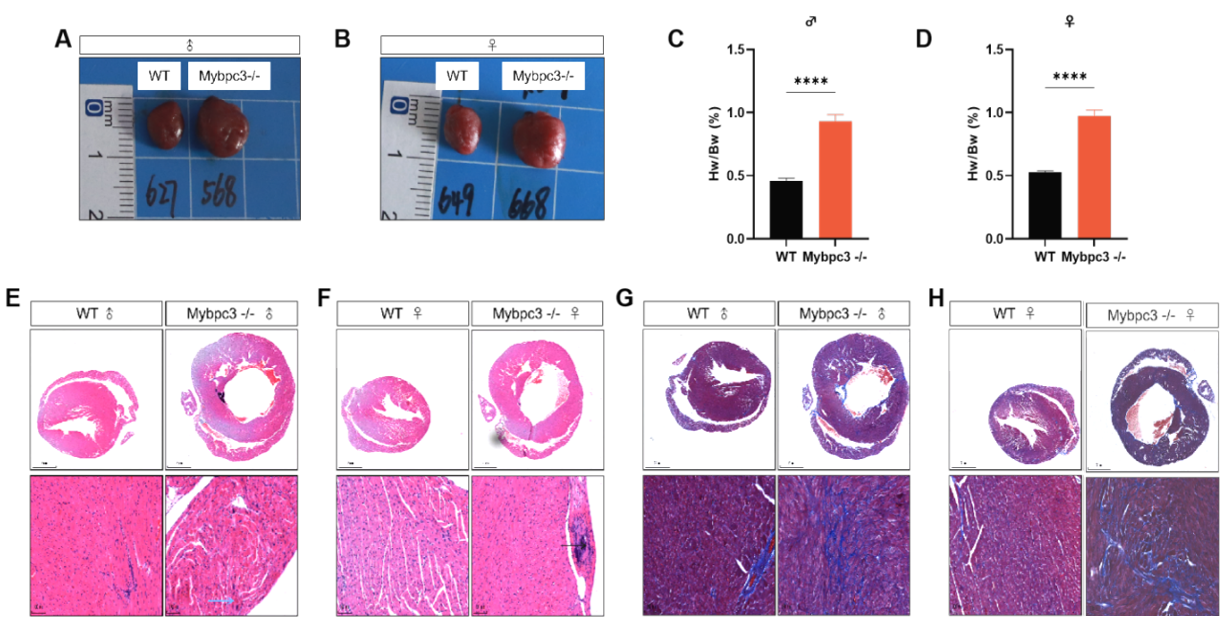
Fig 4. Pathologic analysis of C57BL/6JGpt-Mybpc3em19Cd10922/Gpt hearts 3 months after birth.
C57BL/6JGpt-Mybpc3em19Cd10922/Gpt mice exhibited marked cardiac enlargement accompanied by a significantly elevated heart weight-to-body weight (HW/BW) ratio compared to wild-type controls. Histopathological analysis via H&E and Masson's trichrome staining revealed pronounced cardiomyocyte hypertrophy and extensive myocardial fibrosis in mutant hearts, characterized by increased size of cardiac fibers and collagen deposition, particularly in the perivascular and interstitial regions. These structural abnormalities align with the phenotypic hallmarks of eccentric hypertrophy, reflecting both compensatory adaptive remodeling and progressive pathological decompensation. Notably, the fibrotic lesions exhibited a patchy distribution pattern, suggesting dysregulated extracellular matrix turnover secondary to sustained biomechanical stress in the absence of functional Mybpc3.