
Hypertrophic Cardiomyopathy (HCM) Mouse Model: Myh6 R403Q
Hypertrophic Cardiomyopathy Mouse Model: C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt
Similar to MYH7 in human, in mice, Myh6 encodes the alpha-myosin heavy chain (α-MHC), a key contractile protein in cardiac muscle that drives sarcomere assembly and myocardial contraction by ATP-dependent actin-myosin cross-bridge cycling. Murine Myh6 shares functional homology with human MYH7, as both are predominantly expressed in ventricular cardiomyocytes and critically regulate myocardial contractility. The 404th amino acid residue in murine Myh6 and its surrounding sequences exhibit high homology with the 403rd residue region of human MYH7. Structural studies confirm that this mutation disrupts the binding affinity between myosin heavy chain and cardiac myosin-binding protein C (cMyBP-C), mirroring the pathogenesis of human MYH7 R403Q variants and recapitulating human hypertrophic cardiomyopathy pathology.
Strategy
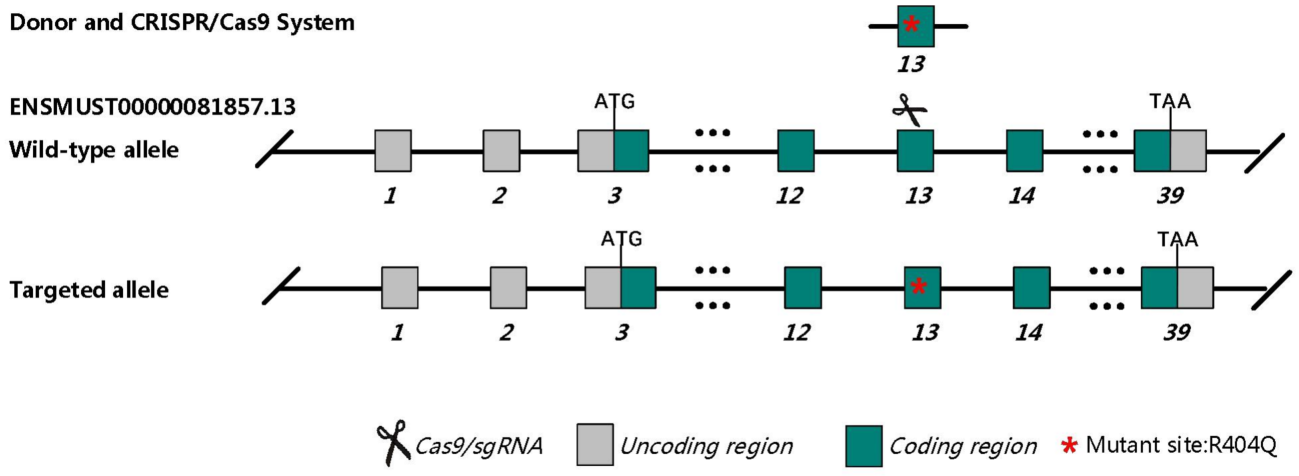
Fig.1 Schematic diagram of C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt model strategy.
C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt mice displayed eccentric hypertrophy cardiomyopathy after birth 4 months.
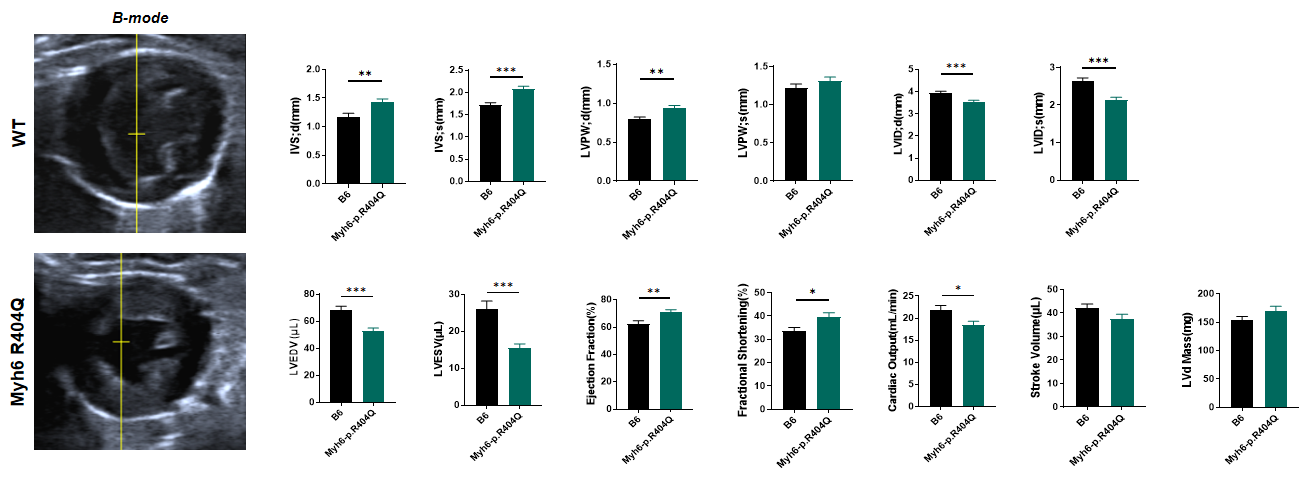
Fig 2. Echocardiography of C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt mice 4 months after birth.
Echocardiographic analysis revealed significant cardiac hypertrophy in Myh6 R404Q+/− mice compared to wild-type (WT) controls. The mutant hearts demonstrated significant increases in both interventricular septal thickness at end-diastole (IVSd) and left ventricular posterior wall thickness at end-diastole (LVPWd) but decreased left ventricular end-diastolic volume (LVEDV) and end-systolic volume (LVESV), coupled with slightly increased ejection fraction (EF) and fractional shortening (FS). These structural and functional aberrations establish that Myh6 mutation is causally linked to the development of concentric hypertrophic cardiomyopathy, highly recapitulating features of human Myh6-associated cardiomyopathy. n=10 per group, all data represented as MEAN ± SEM.****P<0.0001,by t-test. n=10 per group, all data represented as MEAN ± SEM.**P<0.01, ***P<0.001, ****P<0.0001,by t-test.
C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt mice display severe eccentric hypertrophic cardiomyopathy 8 months after birth.
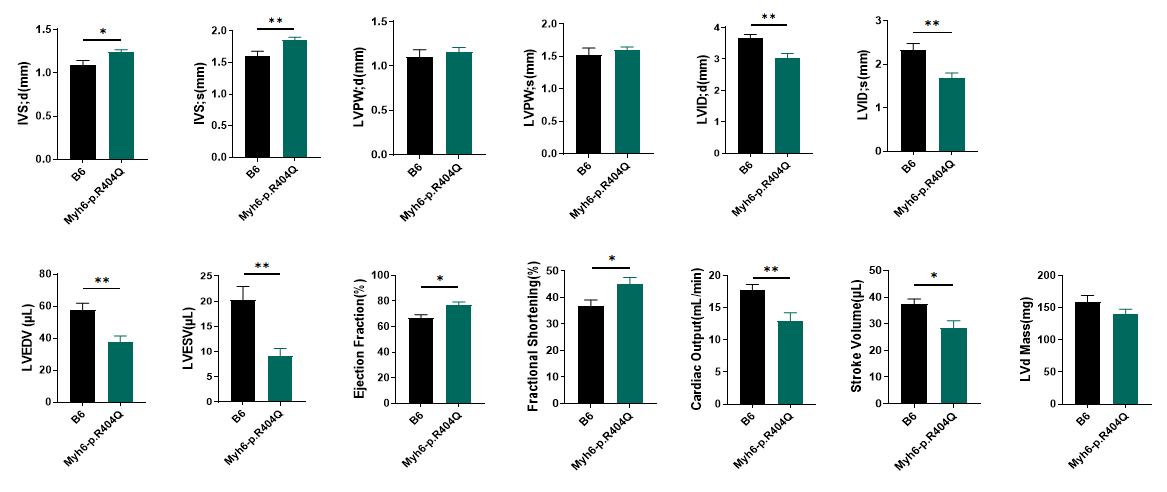
Fig 3. Echocardiography of C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt mice after birth 8 months.
Myh6 R404Q+/− mice survive for up to 8 months, during which concentric myocardial hypertrophy progresses to severe pathology. Beyond ventricular wall thickening and reduced chamber dimensions, mutant hearts exhibit significantly diminished stroke volume (SV) and cardiac output (CO), indicating transition to systolic dysfunction and early-stage heart failure. These hemodynamic impairments correlate with preserved ejection fraction but markedly elevated ventricular filling pressures, recapitulating clinical hallmarks of advanced hypertrophic cardiomyopathy. Notably, the temporal progression of hypertrophy-to-failure in this model parallels human MYH7 R403Q cardiomyopathy timelines, underscoring its translational relevance for studying disease mechanisms and therapeutic interventions. *P<0.05, **P<0.01 by t-test.
Sarcomeric disorganization and cardiac remodeling in C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt mice.
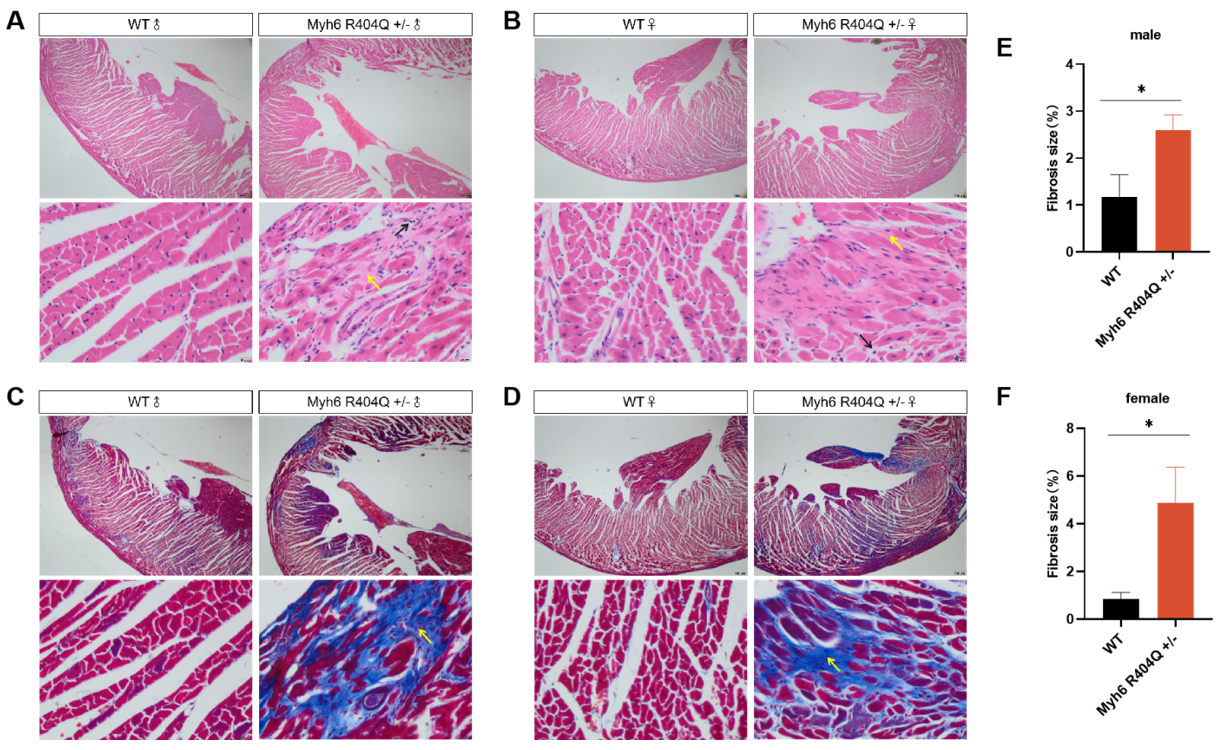
Fig 4. Pathologic analysis of C57BL/6JGpt-Myh6em1Cin(R404Q)/Gpt hearts 8 months after birth.
Histological evidence showed Myh6 R404Q+/− hearts exhibit extensive interstitial and perivascular fibrosis (blue/green collagen deposition in trichrome/sirius red-stained sections) compared to WT, with disrupted cardiomyocyte architecture. Fibrotic lesions are distributed across ventricular walls, suggesting progressive extracellular matrix remodeling, a hallmark of hypertrophic cardiomyopathy (HCM) progression. Quantitative Sex Disparity displayed mutants show a 2.5-fold increase in fibrosis area in male and 3-fold elevation in female, suggesting estrogen-independent fibrotic susceptibility. Elevated fibrosis correlates with diastolic stiffness and arrhythmogenic substrates, aligning with prior observations of impaired cardiac output and hypertrophy in this model. *P<0.05 by t-test.
MYK-461 (Mavacamten) induces transient myocardial relaxation in Myh6 R404Q mutant hearts.
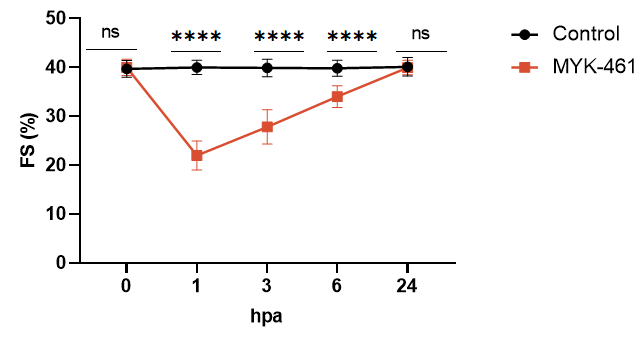
Fig 5. Myh6 R404Q mice respond to standard-of-care treatment. CAMZYOS® (mavacamten), the first-in-class cardiac myosin inhibitor approved by the U.S. FDA, is specifically indicated for symptomatic adults with obstructive hypertrophic cardiomyopathy (oHCM) classified as New York Heart Association (NYHA) class II-III. This novel therapeutic agent selectively modulates β-cardiac myosin ATPase activity, reducing excessive actin-myosin cross-bridge formation and shifting myocardial contractility toward an energy-efficient "super-relaxed" state. Preclinical validation in the Myh6 R404Q/+ murine model (a phenocopy of human MYH7 R403Q cardiomyopathy) demonstrated rapid pharmacodynamic effects. A single dose of MYK-461 (mavacamten's preclinical code) at 10 mg/kg significantly decreased fractional shortening (FS) by 18% within 24 hours compared to vehicle-treated R404Q/+ controls (*p<0.05). This acute reduction in systolic function aligns with mavacamten's mechanism of action, which directly targets sarcomeric hypercontractility, a hallmark of HCM pathophysiology. ns, no significance; ****P<0.0001,by t-test.